popgen-evomics
1. Look at the data and get familiar with them
The data consist of genomic data from chromosome 15 for all the samples in the Year1 release of the HPRC and it is stored on the server of the course at this address:
/home/genomics/workshop_materials/population_genomics/
Data is in the Variant Call Format (VCF), a standard file format used in bioinformatics for storing gene sequence variations. More information about the VCF format can be found here. Within the same folder, you will also find the index file for the VCF file with the extension .tbi
ll /home/genomics/workshop_materials/population_genomics/
-r--r--r-- 1 genomics workshop 253M Dec 20 13:08 chr15.pan.fa.a2fb268.4030258.6a1ecc2.smooth.reliable.vcf.gz
-r--r--r-- 1 genomics workshop 65K Dec 20 13:08 chr15.pan.fa.a2fb268.4030258.6a1ecc2.smooth.reliable.vcf.gz.tbi
zcat /home/genomics/workshop_materials/population_genomics/chr15.pan.fa.a2fb268.4030258.6a1ecc2.smooth.reliable.vcf.gz | less -S
Look at the file name, notice the chromosome name chm13#chr15: it indicates that the vcf that we will use was build using CHM13 as a reference, i.e. the genomic coordinates are based on the sequence of CHM13
CHM13 is a human genome reference. It represents a significant advancement in the field of genomics, providing a more complete and accurate representation of the human genome. CHM13 includes regions that were previously difficult to sequence and assemble, making it a valuable resource for a wide range of genetic research. This reference genome helps in better understanding human genetic diversity, disease mechanisms, and evolutionary biology. It is particularly useful in studies where precise genomic information is crucial.
CHM13 is derived from a hydatidiform mole, which is a rare growth that forms inside the womb at the beginning of a pregnancy. A hydatidiform mole contains a near-complete set of human chromosomes, but with two copies from the father and none from the mother. This unique genetic makeup, with only paternal chromosomes and no maternal contribution, makes it an ideal biological source for sequencing to create a reference human genome. The lack of maternal chromosomes results in a homozygous genetic profile, simplifying the assembly and analysis of the genome. This characteristic of CHM13 has been leveraged to create a more accurate and complete reference human genome.
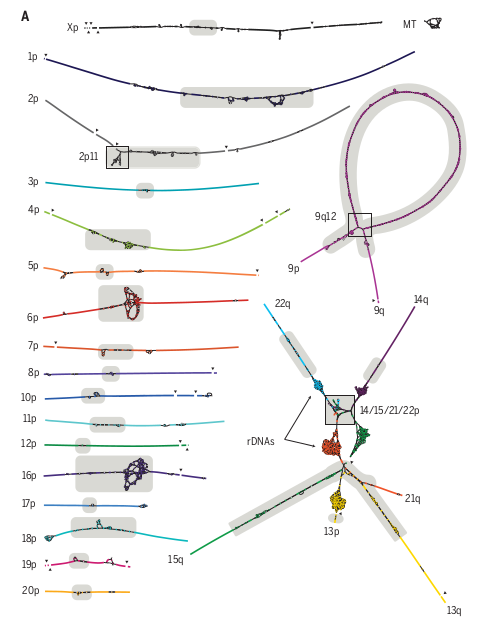
Fig. 2. from (PMID: 35357919) High-resolution assembly string graph of the CHM13 genome. (A) Bandage (60) visualization, where nodes represent unambiguously assembled sequences scaled by length and edges correspond to the overlaps between node sequences. Each chromosome is both colored and numbered on the short (p) arm. Long (q) arms are labeled where unclear. The five acrocentric chromosomes (bottom right) are connected owing to similarity between their short arms, and the rDNA arrays form five dense tangles because of their high copy number. The graph is partially fragmented because of HiFi coverage dropout surrounding GA-rich sequence (black triangles). Centromeric satellites (30) are the source of most ambiguity in the graph (gray highlights). MT, mitochondria.
Explore the vcf using the query option of bcftools a toolset for variant calling and manipulating VCF (Variant Call Format) and BCF (Binary Call Format) files, including tasks like viewing, merging, and querying the data. Look at the query manual page.
Look at the samples included in the dataset:
bcftools query -l /home/genomics/workshop_materials/population_genomics/chr15.pan.fa.a2fb268.4030258.6a1ecc2.smooth.reliable.vcf.gz
## query extracts fields from VCF or BCF files and outputs them in user-defined format.
## -l list sample names and exit
This command wil list the samples, note a sample called grch38, this is the human reference sequence GRC-hg38, currently widely used and annotated.
2. Clone the repository
While it is possible to access and follow the tutorial directly from the web, having all the code available locally might be better, especially if you intend to run the R scripts. The tutorial is hosted on GitHub, therefore you can clone the entire folder to your local machine for easier access and execution of the scripts.
Clone the repository and spend some time navigating the folder to get familiar with the content:
cd ~/workshop_materials/population_genomics
git clone https://github.com/ColonnaLab/popgen-evomics.git
Now look at the metadata, e.g. the information relative to samples, a set of files with info on the geographical origin of the samples in the vcf, take two minutes to open the files and look at them:
cd popgen-evomics
cd metadata
3. Organize your workspace
Organizing your workspace efficiently is crucial. While you have the flexibility to arrange it according to your preferences, it’s recommended to adopt the filesystem structure outlined here. This approach facilitates easier sharing of information and more effective debugging of problems.
Make a folder called mytutorial within the evomics folder that you just cloned
cd popgen-evomics/
mkdir mytutorial
Make subfolders for each session of the tutorial:
cd mytutorial
mkdir data
mkdir 1_afs
mkdir 2_fst
mkdir 3_pca
4. Convert vcf data to plink2 file format
Archaeology of File Formats: plink was developed before the creation of the vcf format and originally read genetic data from a set of files in its specific format. Over time, both the plink file format and the software itself evolved, with plink gaining the capability to read vcf files. In this step we are going to learn about file conversion for you to have a reference for this command if you need it in the future-
For certain analyses in this course, we will use plink2, a powerful tool for genetic data analysis. Although plink2 is capable of reading data directly from VCF files, understanding how to convert VCF files to PLINK format can be useful.
cd data
plink2 --vcf /home/genomics/workshop_materials/population_genomics/chr15.pan.fa.a2fb268.4030258.6a1ecc2.smooth.reliable.vcf.gz \
--make-pgen \
--allow-extra-chr \
--vcf-half-call m \
--out chr15
## --vcf loads a genotype VCF file
## --make-pgen creates a new PLINK 2 binary files
## --allow-extra-chr force to accept chromosome code 'chm13#chr15'
## --vcf-half-call specify how genotype half-call should be processed.
# The current VCF standard does not specify how '0/.' and similar GT (genotype) values should be interpreted.\
# By default (mode 'error'/'e'), PLINK errors out and reports the line number of the anomaly.\
# Should the half-call be intentional, though (this can be the case with Complete Genomics data), you can request the following other modes:
# 'haploid'/'h': Treat half-calls as haploid/homozygous (the PLINK 2 file format does not distinguish between the two)
# 'missing'/'m': Treat half-calls as missing.
## --out gives a human-readable name
This command will produce this fileset, all named chr15, but with different file extensions:
-rw-rw-r-- 1 enza enza 906 gen 16 09:33 chr15.log
-rw-rw-r-- 1 enza enza 9499035 gen 16 09:33 chr15.pgen
-rw-rw-r-- 1 enza enza 503 gen 16 09:33 chr15.psam
-rw-rw-r-- 1 enza enza 1233336557 gen 16 09:33 chr15.pvar
Open the chr15.log file and read it to check for error messages (if any) or just to make sure all worked well:
cat chr15.log
Plink1 and Plink2
plink is one of the most popular software for genomic data analysis.
Latest version plink2 accepts pfile binary format and suports multiallelic sites.
Previous version plink1 accepts bfile binary format.
This is a reference to plink2 file formats.